
科技改變生活 · 科技引領未來
科技改變生活 · 科技引領未來

(報告出品方/作者:國金證券,王班)專注于AR相關疾病的新藥研發公司開拓藥業是專注于自主研發同類首創及同類最佳的腫瘤藥物及雄激素受體(AR)相關疾病藥物的新藥研發公司,目前已有7款臨床階段的在研產品。AR拮抗劑普克魯胺正在開展治療COVID
(報告出品方/作者:國金證券,王班)
專注于 AR 相關疾病的新藥研發公司
開拓藥業是專注于自主研發同類首創及同類最佳的腫瘤藥物 及雄激素受體(AR)相關疾病藥物的新藥研發公司,目前已有 7 款臨床階 段的在研產品。 AR 拮抗劑普克魯胺正在開展治療 COVID-19 的三項國際 多中心臨床試驗,以及治療前列腺的中國 III 期臨床試驗和美國 II 期臨床試 驗。外用 AR 拮抗劑福瑞他恩正在開展針對雄激素性脫發和痤瘡的臨床試 驗,其中治療男性雄激素脫發的中國臨床已經進入 III 期。ALK-1 抑制劑 GT90001 聯合 PD-1 治療肝癌也已經進入 II 期臨床。此外,mTOR 多激酶 抑制劑迪拓賽普、Hedgehog/SMO 抑制劑 GT1708F、AR-PROTAC 化合 物 GT20029、以及 PD-L1/TGF-β雙抗 GT90008 等處于臨床早期階段, 還有多款臨床前階段的候選藥物。
公司的蘇州工廠已于 2020 年獲得生產許可證,后續將完成中國 GMP 達標、 以及北美和歐盟 GMP 認證。蘇州工廠計劃用于普克魯胺片劑生產及福瑞 他恩臨床藥品自主生產,預計 2022 年普克魯胺產能將達到 5000 萬人份以 上(每人份 28 片)。2021 年 4 月,公司與華益泰康就擴大普克魯胺產能達 成戰略合作協議,華益泰康具有美國 FDA 認證經驗,將有利于加速普克魯 胺的生產和全球商業化進程。
公司也積極拓展商業化合作。就普克魯胺的新冠適應癥,公司與復星醫藥 達成了在印度和 28 個非洲國家的商業化合作協議,與印度尼西亞公司 Etana 達成了在印度尼西亞的商業化合作協議。此外,公司還與上藥控股、 京東大藥房、國藥集團達成了商業化合作。(報告來源:未來智庫)
普克魯胺:完成治療輕中癥新冠患者的全球多中心 III 期臨床
新冠小分子藥物優勢
小分子藥物則主要作用于病毒進入細胞以及在細胞內復制的過程。病毒進 入細胞需要跟細胞表面的 ACE2 受體結合,調控 ACE2 受體的表達能夠降 低病毒進入細胞的機會,從而減少病毒在細胞內的復制。3CL 蛋白酶負責 將病毒遺傳物質翻譯出來的多蛋白鏈水解成能發揮作用的單個功能蛋白, 3CL 蛋白酶抑制劑能夠抑制 3CL 蛋白酶的作用,從而阻止病毒后續的一系 列復制活動。RNA 依賴的 RNA 聚合酶(RdRp)是病毒 RNA 復制過程中 的聚合酶,RdRp 抑制劑抑制 RdRp 的作用,能干擾或抑制病毒的復制。
小分子藥物具有對變異株普遍有效的潛力。新冠病毒不斷發生變異的過程 中,表面結構蛋白很容易發生結構的改變,但是胞內過程相對保守,不易 發生突變,因此作用于胞內過程的小分子藥物具有對變異株普遍有效的潛 力。相比之下,新冠疫苗和新冠中和抗體大多作用于新冠病毒表面結構蛋 白,效果容易受到病毒變異的影響。曾經在美國獲得緊急使用授權 (Emergency Use Authority, EUA)的多個中和抗體均因為對變異株無效, 目前在美國已經暫停使用。
小分子藥物具有便利性優勢、價格優勢、產能優勢。(1)便利性優勢:大 部分小分子藥物可以做成口服劑型,相對于需要靜脈輸液的中和抗體,具 有便利性優勢,尤其是無需住院的輕中癥患者,可以在確診后自己口服藥 物,不會對醫療資源(如醫院床位資源、醫護人員資源等)造成額外負擔。價格優勢:目前在美國,中和抗體類治療藥物的價格約為 2000 美元/療程, 而默沙東小分子口服藥物 Molnupiravir 與美國政府的訂單價格約為 700 美 元/療程,輝瑞小分子口服藥 Paxlovid 與美國政府訂單價格約為 529 美元/ 療程;(3)產能優勢,小分子口服藥物的生產與大分子生物藥相比相對簡 單,生產線和產能提升相對容易,能滿足更多患者的需求。因此,我們更 看好小分子藥物在新冠治療中的應用前景。
新冠小分子藥物競爭格局
目前,全球已有 3 款小分子藥物獲得上市或 EUA 批準,分別是輝瑞的小分 子口服藥 Paxlovid(奈瑪特韋+利托那韋)、默沙東的小分子口服藥 Lagevrio(molnupiravir)、和吉利德的小分子藥物瑞德西韋注射劑。
目前這三種藥物均獲批終于治療輕中癥、高風險患者,即目前病情不需要 住院、但是伴隨進展到住院的高風險因素(包括年齡在 65 歲以上,或有 基礎疾病)。在臨床試驗中,針對輕中癥、高風險患者,Paxlovid 降低了 88%-89%的住院率,瑞德西韋降低了 87%的住院率,Molnupiravir 降低 了 30%的住院率。
而針對大量的輕中癥、一般風險的患者,目前暫無新冠特效藥獲批。
目前處于研發后期的小分子藥物包括開拓藥業的普克魯胺、鹽野義的 S217622、君實生物和旺山旺水合作開發的 VV116、以及真實生物的阿茲夫 定。其中普克魯胺已經完成一項全球多中心(美國為主)的 III 期臨床,鹽 野義完成了日本 II 期并啟動了全球 III 期,VV116 在烏茲別克斯坦獲得 EUA 授權、同時正在進行兩項全球多中心的 III 期臨床,阿茲夫定處于 III 期臨床后期。同時,國內還有多家企業布局新冠小分子藥物的研發,目前 處于研發早期階段。
普克魯胺治療新冠作用機理
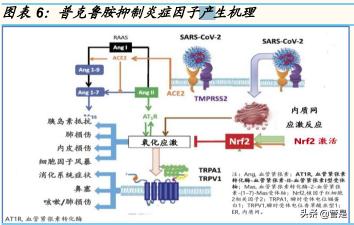
普克魯胺是公司研發的雄激素受體(AR)拮抗劑。普克魯胺通過降低 ACE2 和 TMPRSS2 的表達,抑制新冠病毒侵入宿主細胞,同時通過激活 Nrf2 通路抑制炎癥因子產生,下調巨噬細胞中的 iNOS 抑制細胞因子風暴 的發生,從而降低中重度新冠患者嚴重程度。
普克魯胺抑制新冠病毒侵入宿主細胞。新冠病毒利用 TMPRSS2 和 ACE2 介導的雙重啟動作用,引發病毒刺突蛋白水解,進而幫助病毒 RNA 侵入宿 主細胞。而 ACE2 和 TMPRSS2 受到人體雄激素受體(AR)信號通路的正向 調控。普克魯胺靶向 AR-ACE2/TMPRSS2 信號軸,通過抑制 ACE2 和 TMPRSS2 的表達來降低或阻斷 COVID-19 進入宿主細胞, 從而在源頭上 降低新冠病毒感染的可能性。
普克魯胺抑制炎癥因子產生和細胞因子風暴的發生,從而降低中重度新冠 患者嚴重程度。新冠肺炎患者的一個共同點是氧化還原內穩態轉化為了氧 化應激,即活性氧 (ROS)的積累;新冠病毒結合 ACE2,而 ACE2 下調增 強 AT1R 軸并產生氧化應激;而 Nrf2 信號的上調能夠抑制白細胞介素-6 (IL-6)、促炎細胞因子、以及趨化因子的過度產生;同時,Nrf2 也抑制了涉 及氧化應激的核因子κB (NF?B)的激活;激活 Nrf2 可抑制新冠肺炎感染中 后期多種炎癥因子的產生,從而可降低中重度新冠患者嚴重程度。另一方 面,新冠患者感染嚴重程度與誘導型一氧化氮合酶(iNOS)介導的細胞風暴 密切相關。普克魯胺能下調巨噬細胞極化/活化標記物—誘導型一氧化氮合 酶(iNOS),這表明普克魯胺能抑制新冠患者 M1 型巨噬細胞介導的細胞因 子風暴的產生,從而降低新冠患者的疾病嚴重程度。
普克魯胺全球多中心注冊性臨床頂線數據讀出
目前,公司正在開展普克魯胺治療新冠的 3 項全球多中心的注冊性 III 期臨 床試驗,包括一項以美國患者為主的全球多中心輕中癥 III 臨床、一項中國 參與的全球多中心輕中癥 III期臨床、以及一項全球多中心的重癥 III期臨床。
2022 年 4 月 6 日,公司公布了美國患者為主的全球多中心 III 期臨床試驗 的頂線數據。
這項臨床共納入了 733 例患者,以美國患者為主。這項試驗沒有對患者人 群進行風險因素的篩選,所有 3 天內確診、不需要住院的成人患者均可以 入組。試驗采取 1:1 隨機分組,治療組接受普克魯胺和標準治療,對照組 接受安慰劑和標準治療,普克魯胺的用藥方式為 200mg 一次,每天一次口 服,治療時間為 14 天。在完成 14 天治療后,患者會接受 28 天隨訪。試 驗的主要臨床終點為第 28 天未發生住院或吸氧或死亡的受試者百分比,次 要臨床終點為 28 天內受試者住院或吸氧或死亡的發生率、第 7 天、第 14 天和第 28 天臨床評估、病毒載量變化、安全性評估等。
試驗結果顯示,普克魯胺有效降低住院率:
在服藥至少一天的 730 例受試者中,對照組有 8 例患者住院,治療組 有 4 例患者住院,普克魯胺降低了 50%的住院率,未披露統計學顯著 性;
在服藥大于一天的 721 例受試者中,對照組有 7 例患者住院,治療組 有 2 例患者住院,普克魯胺降低了 71%的住院率,未披露統計學顯著 性;
在服藥大于七天的 693 例受試者中,對照組有 6 例患者住院,治療組 沒有患者住院,普克魯胺降低了 100%的住院率,具有統計學顯著性。
此外,在伴有高風險因素的受試者中,治療組沒有患者住院,普克魯 胺降低了 100%住院率,具有統計學顯著性。
針對次要終點,普克魯胺能顯著降低病毒載量,并能改善新冠肺炎的 部分相關癥狀。
安全性結果顯示,普克魯胺整體耐受性良好。
公司仍需要 4-6 周進行詳細的數據分析,預計 5 月將披露更詳細的數據, 并計劃提交美國、中國及其他國家的上市申請。
2021 年 12 月 27 日公司披露了這項臨床的中期數據,基于 348 人的數據 分析顯示,臨床終點沒有達到統計學顯著性。我們認為重要的影響因素有 兩個:
納入了所有輕中癥人群,導致住院率整體降低。輝瑞和默沙東的新冠 口服藥均只納入了高風險人群,相對來說整體住院率會較高;開拓藥 業的普克魯胺臨床納入了全部輕中癥人群,整體住院率就會偏低。
樣本量不足,導致住院事件數太少。同樣是中期分析,輝瑞納入了 1219 例高風險輕中癥患者,默沙東納入了 755 例高風險輕中癥患者, 普克魯胺的中期分析僅納入了 348 例輕中癥患者,其中還包含非高風 險的患者,導致整體住院事件數太少。
在本次披露的最終數據中,包含了 730 例輕中癥患者,樣本量的放大使得 住院事件數上升,因此在服藥 7 天以上人群及高風險人群中達到了統計學 顯著性。
但是,在該項臨床試驗的所有入組人群中,普克魯胺降低住院率的主要終 點尚未披露統計學顯著性,存在不具有統計學顯著性,即未達到主要臨床 終點的風險,可能會對普克魯胺是否獲得上市或 EUA 批準造成影響。
此前 FDA 曾基于亞組數據批準巴瑞替尼的 EUA。巴瑞替尼曾經開展一項 治療新冠住院患者的 COV-BARRIER 試驗,在全部入組的 1518 例住院患 者中,巴瑞替尼沒有達到降低死亡率的主要臨床終點,但是隨后的亞組分 析顯示,在 101 例需要機械通氣或 ECMO 的患者中,巴瑞替尼顯著降低了 死亡率。基于這項亞組分析,巴瑞替尼于 2021 年 7 月獲得了 FDA 的 EUA 授權,用于治療 2 歲以上的需要補充氧氣或機械通氣或 ECMO 的新冠住院 患者。目前,巴瑞替尼仍舊被美國 NIH 指南推薦用于治療需要補充氧氣或 機械通氣的新冠住院患者。
我們認為,基于目前披露的這項 III 期臨床的頂線數據,普克魯胺有機會獲 得美國和/或中國、及其他國家的上市批準或 EUA批準。
此外,公司另外兩項 III 期臨床試驗也在進行中。其中,中國參與的全球多 中心輕中癥 III 期臨床中,僅納入了高風險的輕中癥患者,未來有望獲得更 多普克魯胺的療效數據。針對重癥患者的全球多中心 III 期臨床有望支持普 克魯胺未來獲批治療新冠重癥患者的適應癥。
普克魯胺巴西研究者發起臨床數據優異
此前, 在巴西開展的 3 項研究者發起的臨床實驗中,普克魯胺治療住院患 者和非住院患者都已經取得了積極的臨床數據。
在巴西開展的針對非住院患者的臨床中,普克魯胺降低了 90%的住院率。 針對非住院的輕中癥患者,巴西研究者發起了兩項臨床試驗,分別針對男 性患者和女性患者。針對男性患者的研究一共納入了 268 例患者,在第 30 天,普克魯胺組住院率為 2%,而對照組住院率為 26%,普克魯胺住院率 低于對照組 90%以上。同時在輔助吸氧、機械通氣率、高流量氧氣、死亡 率等次要終點也表現出優于安慰劑的臨床結果。針對女性患者的研究,在 2021 年 1 月 7 日公布了 95 名患者(普克魯胺治療組 60 名患者,安慰劑對 照組 35 名患者)的中期數據分析,普克魯胺治療組住院率為 1.7%,對照 組住院率為 17.1%,普克魯胺住院率低于對照組 90%,同時在 ICU 比例、 機械通氣率、死亡率等臨床終點的數據也均優于安慰劑對照組。
在巴西開展的針對住院患者的臨床中,普克魯胺降低了 78%的死亡風險。 針對住院患者的巴西臨床試驗一共招募了 645 例住院患者,包括男性患者 和女性患者,患者 1:1 隨機進入普克魯胺治療組或安慰劑對照組。在第 14 天,普克魯胺治療組的康復出院率比安慰劑對照組增加了 128%;在第 28 天,普克魯胺治療組的住院患者死亡風險比安慰劑對照組降低了 78%,住 院至康復時間縮短了 5 天。在安全性方面,普克魯胺治療組的患者發生胃 腸道副作用的比例更高,而在腎衰竭和肝損傷方面的發生率低于安慰劑組, 但是沒有顯著區別。
基于巴西臨床數據,2021 年 7 月,普克魯胺獲得巴拉圭的 EUA 批準,用 于新冠住院患者的治療。
普克魯胺產能及商業化準備充分
按每人每療程 28 片(每片 100mg)計算,公司目前具備每月生產 100 萬 人份普克魯胺的產能,預計今年底將達到每年生產 5000 萬人份的產能。 同時,公司就擴大產能與華益泰康達成合作,借助華益泰康的 FDA 認證經 驗,公司有望迅速獲得 FDA 的 GMP 認證。
商業化方面,公司目前已經在印度、非洲、印尼等海外市場達成了商業化 合作,有望借助成熟藥企的商業化經驗和渠道,打開海外市場。
2021 年 7 月 15 日,公司宣布與上海復星醫藥就普克魯胺治療新冠在 印度和 28 個非洲國家的商業化達成合作協議,雙方將共同推進普克魯 胺新冠適應癥的 EUA 申請、推廣和銷售工作。根據協議條款,復星醫 藥產業將獲得普克魯胺在合作區域的獨家注冊和商業化銷售權益,并 就此支付公司不超過人民幣 5.6 億元款項,包含首付款、開發里程碑 約人民幣 1.1 億元以及商業化里程碑不超過人民幣 4.5 億元。此外,公 司將基于普克魯胺在合作區域內的未來凈銷售額,分級收取不低于利 潤總額的 50%作為銷售提成。
2021 年 8 月 25 日,公司宣布與印度尼西亞生物科技公司 Etana Biotechnologies 關于普克魯胺治療新冠在印度尼西亞的商業化達成合 作協議。
普克魯胺治療新冠銷售預測
上市成功率及時間假設:普克魯胺美國 III 期臨床已經完成,預計將于 1-2 個月后提交上市申請。基于頂線數據分析,假設獲得歐美市場 EUA 上市或 EUA 批準的成功率為 40%,和其他海外市場 EUA 批準的成功率為 50%, 獲得中國上市批準的成功率為 70%,假設獲批上市時間為 2022 年下半年
定價假設:對于美國市場,考慮到默沙東 Molnupiravir 定價為 700 美元/療 程,輝瑞 Paxcovid 定價約為 530 美元/療程,我們假設普克魯胺在美國定 價為 1500 人民幣/療程,即 210 美元/療程左右,每年降價 2%;在中國及 其他新興市場定價為 1000 人民幣/療程,每年降價 2%;
新冠發病率假設:
歐美市場:美國在 2020 年和 2021 年的新冠發病率分別為 6.0%和 10.6%左右,2022 年初受 Omicron 變異株影響新增病例數暴發,保守 預計 2022 年發病率約為 18%,預計 2023 年往后,發病率逐漸下降至 6%的穩定水平。歐洲市場參考美國市場發病率。
中國:中國 2020 年和 2021 年的新冠確診人數分別為 9.4 萬人和 2.1 萬人,2022 年初國內疫情反復,已有約 30 萬新增陽性病例(含無癥 狀感染者),預計 2022 年感染人數可能超過 40 萬人。假設 2023 年起, 疫情平穩發展,最終流感化。根據疾病預防控制局數據,2021 年 2 月 至 2022 年 2 月國內累計流感患者約 95.5 萬人,初步假設到 2026 年 以后,每年新冠患者也在 100 萬人左右,發病率在 0.08%。
新興市場:公司與復星醫藥及 Etana 公司合作范圍涉及印度、非洲、 印尼等新興市場,假設普克魯胺將來有潛力獲批的新興市場總人口為 20 億人。目前全球平均發病率約為 7%,新興市場部分地區醫療條件 受限,診斷率偏低,假設發病率穩定在為 3%。
普克魯胺市場份額假設:
美國:目前 FDA 會批準的適應癥人群還存在不確定性,假設普克魯胺 2022 年獲批適用于所有輕中癥患者(96%的患者);在一般風險患者 (71%的患者)中,目前暫無新冠特效藥獲批,假設 5%的一般風險患 者會使用普克魯胺;在高風險患者(25%的患者)中,目前已有輝瑞 Paxlovid、吉利德瑞德西韋、默沙東 Molnupiravir、禮來中和抗體獲批 使用,假設普克魯胺市場滲透率為 2%;2022 年下半年上市,假設 2022 年滲透率減半。
中國:假設普克魯胺獲批適用于所有輕中癥患者(96%的患者);在一 般風險患者中,大部分患者可能會使用中藥,假設普克魯胺滲透率為 10%;在高風險患者中,國內目前有 Paxlovid 和騰盛博藥中和抗體獲 批,還有大量在研藥物,假設普克魯胺占比為為 5%。此外,2022 年 假設政府備貨 50 萬,后續政府備貨逐漸下降。
新興市場:假設普克魯胺適用于 96%的患者,在適用患者中,考慮到 新興市場有輝瑞 Paxlovid 仿制藥,假設普克魯胺市場份額為 2%。
營收假設:
美國及新興市場:考慮到新冠治療藥物目前處于高需求狀態,銷售以 政府訂單為主,銷售費用較低,我們假設 2022 年普克魯胺在海外市場 的凈利率為 80%,隨著上市產品增多、市場自由化,凈利潤逐漸降低 至 40%。參考公司與復星的合作條款,假設公司在海外市場的營收為 普克魯胺凈利潤的 50%;此外公司還有供貨收入,假設普克魯胺供貨 價為銷售價格的 10%。
中國市場:假設普克魯胺銷售額即為公司營收。
綜合以上假設,我們預計普克魯胺在 2022 年至 2024 年在全球范圍的經風 險調整后的營收分別是 16.3 億元、22.9 億元、15.4 億元,并隨著新冠疫 情常態化發展,逐步下降至 2030 年 6 億元。(報告來源:未來智庫)
普克魯胺治療 AR 陽性腫瘤
中國前列腺癌患者數量多,增速快
前列腺癌是男性人口最常見的癌癥類型之一。前列腺癌始發于前列腺中的 健康細胞發生變化并且失去控制,最終發展成腫瘤。可能導致前列腺癌的 風險因素包括:BRCA1 及/或 BRCA2 基因的突變,其他遺傳變化(HPC1、 HPC2、HPCX、CAPB、ATM 及 FANCA),及家族史及飲食習慣。
根據前列腺癌的發展階段,可以將前列腺癌分為局限性前列腺癌、轉移性 激素敏感性前列腺癌(mHSPC)、和轉移性去勢抵抗性前列腺癌 mCRPC)。
中國前列腺癌發病率呈持續上升趨勢,且晚期比例高,嚴重危害中國男性 健康。2020 年全球前列腺癌發病 141.43 萬例,死亡 37.53 萬例。中國前 列腺患者總數從 2014 年的 8.3 萬人增長到 2018 年的 28.91 萬人,復合年 增長率 36.6%。預計到 2028 年,中國前列腺癌患者將增加至 171.63 萬人。 中國 mCRPC 患者總數從 2014 年的 2.63 萬人增加到 2018 年的 8.14 萬人, 預計 2028 年將增加至 33.23 萬人。美國前列腺癌發病率比中國更高,增 長相對平穩。美國前列腺癌患者人數從 2014 年的 293 萬人增長到 2018 年 的 354 萬人,復合年增長率 2.6%。預計到 2028 年,美國前列腺癌患者將 增加至 518 萬人。美國 mCRPC 患者從 2014 年的 30.96 萬人增加到 2018 年的 33.84 萬人,預計 2028 年將增加至 37.42 萬人。
根據 Globocan 預測,中國每年新增前列腺癌患者將從 2020 年的 11.5 萬 人,增長至 2030 年的 17.0 萬人,年復合增長率接近 4%。美國每年新增 前列腺癌患者將從 2020 年的 21.0 萬人,增長至 2030 年的 24.0 萬人,年 復合增長率為 1.4%。
中國 AR 藥物競爭格局
根據 2021 年 CSCO 前列腺癌診療指南,對于局限性前列腺癌患者,推薦 前列腺癌根治術及外照射放療(External irradiation radiotherapy,EBRT) 或近距離放療等治療手段,中危及高危患者同時推薦雄激素剝奪治療 (Androgen deprivation therapy,ADT,也稱為去勢療法)。轉移性激素敏 感性前列腺癌患者仍然對去勢療法敏感,對于這個階段的患者,推薦以 ADT 治療為基礎,根據情況聯合阿比特龍、AR 拮抗劑、激素治療、化療 等治療方案。其中,一代 AR 拮抗劑比卡魯胺和二代 AR 拮抗劑恩雜魯胺、 阿帕他胺均被納入推薦方案。去勢抵抗性前列腺癌患者對于去勢療法已經 產生抵抗,根據既往是否接受過新型內分泌治療和/或化療。其中,AR 拮抗劑恩雜魯胺被推薦為既往未經新型內分泌治療患 者(包括未經化療或多西他賽化療失敗的患者)的首選治療方案,以及既 往新型內分泌治療失敗且未經化療的患者的 III級推薦方案。
自 2019 年以來,中國陸續批準了 3 個 AR 藥物,分別是安斯泰來的恩雜魯 胺,楊森的阿帕他胺,和拜耳的達羅他胺。其中阿帕他胺獲批了 CRPC 和 HSPC 兩類適應癥, 恩雜魯胺和達羅他胺獲批適應癥為 CRPC,HSPC 均處 于臨床研發階段。
最早上市的恩雜魯胺已經于 2020 年底通過醫保談判進入國家醫保乙類, 醫保后年治療費用由 47 萬元下降到 10 萬元左右,降幅 78.7%。此外,豪 森的恩雜魯胺仿制藥也已經獲批上市,年治療費用為 7 萬左右。阿帕他胺 和達羅他胺于 2021 年底進入醫保,年治療費用為 8 萬元至 9 萬元左右。
中國處于臨床階段的 AR 藥物有 5 種,其中公司的普克魯胺、恒瑞的 SHR3680、和海思科的德恩魯胺已進入臨床 III期,處于第一梯隊。
普克魯胺治療前列腺癌研發進展
普克魯胺是治療 mCRPC 的潛在同類最佳藥物。普克魯胺與恩雜魯胺和阿 帕他胺相比,對 AR 配體有更強的親和力,不僅能夠抑制雄激素與 AR 受 體結合,還表現出下調 AR 表達的生物學作用。同時,普克魯胺對 CYP 家 族酶無誘導作用,而恩雜魯胺是 CYP3A4 的強誘導劑,阿帕他胺及其代謝 物是 CYP3A4 和 CYP2C8 的中強誘導劑,這使得普克魯胺具有更廣泛的 聯合用藥的潛力。
在已完成的中國 II 期臨床中,普克魯胺治療 mCRPC 患者顯示出良好的有 效性和安全性。這項 II 期臨床納入了 108 例受試者,受試者均為經標準的 化療方案(含多西紫杉醇的化療方案)治療失敗、或不能耐受或不愿意接 受標準化療治療的 mCRPC 患者。受試者每天接受 100/200/300mg 普克魯 胺治療,28 天為一個周期,直至 6 個周期結束或不耐受。
研究結果顯示,普克魯胺治療組 PSA 下降 50%或更多的患者比例 (PSA50)為 41.9%,其中普克魯胺 200mg/300mg 組的 PSA50 分別達到 45.5%以及 45.7%;ORR 和 SD 達到了 15.8%和 63.2%。截至 2020 年 9 月,普克魯胺平均用藥時長為 10.82 個月,其中,200mg 組平均用藥時長 為 12.02 個月,此外仍有 10 例患者在贈藥階段,反映了潛在的 PFS 時間 可能較長。
安全性方面,普克魯胺整體安全性優于阿比特龍。與恩雜魯胺相比,恩雜 魯胺在其注冊臨床 AFFIRM 中導致的癲癇發生率為 0.9%,在實際使用中 導致癲癇的發生率約為 0.5%,普克魯胺目前臨床試驗中沒有癲癇發生。
目前,普克魯胺有兩項 III 期臨床在中國進行,分別是普克魯胺單藥后線治 療阿比特龍及多西他賽治療失敗的 mCRPC 患者,和普克魯胺聯合阿比特 龍一線治療 mCRPC 患者。兩項臨床將納入總計約 1000 例患者。此外, 普克魯胺還在美國開展一項單藥后線治療阿比特龍及恩雜魯胺治療失敗的 mCPRC 患者的 II期臨床試驗。
普克魯胺也是潛在的乳腺癌治療藥物
乳腺癌是女性最常見的腫瘤,常發生于 50 歲以上女性。在各類乳腺癌患者 中,都有不少比例患者 AR 表達為陽性,因此 AR 也是潛在的乳腺癌治療 靶點。中國 AR+乳腺癌患者從 2014 年的 61.19 萬人增加至 2018 年的 135.60 萬人,預計 2028 年將增至 340.63 萬人。美國 AR+乳腺癌患者從 2014 年的 249.74 萬人增加至 2018 年的 314.37 萬人,預計 2028 年將增 至 501.74 萬人。
目前恩雜魯胺等 AR 拮抗劑已經在美國開展治療 AR 陽性乳腺癌的臨床研 究,普克魯胺是目前唯一在中國開展 AR 陽性轉移性乳腺癌臨床試驗的 AR 藥物。
在已經完成的 I/Ib 期臨床試驗中,普克魯胺耐受程度良好,沒有觀察到劑 量限制性毒性(DLT)及尚未達到最大耐受劑量(MTD);普克魯胺相關的 不良事件被評為 1 級或 2 級。同時普克魯胺對 AR 陽性的患者有較佳的臨 床效果,并在 AR 陽性的三陰乳腺癌患者中顯示出了初步療效。目前,普 克魯胺結合依西美坦、來曲唑、氟維司群治療 HR+和 AR+的轉移性乳腺癌 患者的 Ic 期臨床(CTR20191063)正在進行中,2021 年 8 月 25 日完成 了患者招募。
普克魯胺治療腫瘤銷售預測
普克魯胺二線治療前列腺癌 II 期臨床成功,目前已經處于 III 期臨床中,假 設上市成功率為 70%,2023 年上市;假設上市價格為 24 萬人民幣/年(參 考 AR 進口新藥在國內的上市價格為 28 萬-48 萬/月),2024 年進入醫保后 價格為 7.2 萬人民幣/年(參考 AR 仿制藥醫保后價格約為 7 萬/年),之后 每年下降 2%;假設普克魯胺出廠價為終端價的 70%;二線適用普克魯胺 的患者在 2030 年達到 48%左右(包括一線未使用 AR 的患者,和部分一 線使用 AR 后進展的患者),普克魯胺市場滲透率峰值為 20%;假設患者平 均用藥時間為 II期臨床中的平均用藥時長 12.02 個月;
普克魯胺一線治療前列腺癌目前處于 III 期臨床招募中,暫無 II 期數據,假 設上市成功率為 60%,2024 年上市;普克魯胺藥品價格與二線適應癥一 致;一線患者中 AR 藥物整體滲透率逐年上升,在 2030 年達到 25%,普 克魯胺的市場占有率峰值為 20%;患者的平均用藥時長參考恩扎盧胺在一 線治療前列腺癌的注冊臨床中的平均用藥時長 16.60 個月;
普克魯胺二線治療前列腺癌同時也在美國開展臨床 II 期臨床,考慮到納入 了恩扎盧胺治療失敗的患者,謹慎假設上市成功率為 40%,2025 年上市; 假設美國上市價格為 6 萬人民幣/月(參考恩扎盧胺美國價格約為 9 萬人民 幣/月);假設 AR 藥物在二線患者中整體滲透率為 30%,普克魯胺市場占 有率逐步上升達到 5%;假設患者平均用藥時間為 II 期臨床中的平均用藥 時長 12.02 個月;
普克魯胺治療乳腺癌正在早期臨床試驗中,謹慎假設上市成功率為 30%, 2026 年上市;價格與前列腺癌適應癥一致;AR 在晚期轉移性乳腺癌中滲 透率逐年上升達到 15%,其中普克魯胺市場占有率逐漸從 100%下降到 70%;患者的平均用藥時長參考氟維司群治療 HR 陽性晚期乳腺癌的 PFS 16.60 個月;
綜合以上假設,我們預計普克魯胺治療腫瘤適應癥的銷售峰值約為 13 億元, 經風險調整后的銷售峰值約為 6.0 億元。(報告來源:未來智庫)
福瑞他恩和 GT20029 治療雄激素性脫發和痤瘡
雄激素性脫發市場
雄激素性脫發是頭皮脫發的常見形式,在男性和女性身上都有可能發生。 雄激素性脫發可能在青春期后的任何年齡發生,并且發生率隨年齡增加而 增加。雄激素脫發發生的風險因素包括過度吸煙、家族史、營養不良、壓 力及衰老等。
2018 年,中國有約 9280 萬雄激素性脫發的男性患者,預計 2028 年增長 至 9780 萬人。美國雄激素性脫發的發生率高于中國,2018 年,美國約有 3110 萬雄激素性脫發的患者,預計 2028 年增長至 3390 萬人。
雄激素性脫發的治療藥物目前主要包括米諾地爾和非那雄胺。米諾地爾是 最常用的治療藥物,2018 年中國和美國接受米諾地爾治療的患者比例估計 分別為 70%和 75%,而非那雄胺在中國和美國的患者比例估計為 30%和 25%。米諾地爾產生的副作用包括丙二醇過敏和直立性低血壓,非那雄胺 可能產生的副作用包括性欲減退、性功能障礙及射精障礙。
中國米諾地爾和非那雄胺的銷售額由 2014 年的 10.64 億人民幣增長至 2018 年的 14.70 億人民幣,預計 2028 年中國雄激素性脫發的藥物市場將 增長至 47.33 億人民幣。按 2018 年價格計算,中國米諾地爾及非那雄胺 的年治療費用分別為 199 美元及 314 美元。美國米諾地爾和非那雄胺的銷 售額由 2014 年的 3.69 億美元增長至 2018 年的 4.08 億美元,預計 2028 年美國雄激素性脫發的藥物市場將增長至 14.18 美元。按 2018 年價格計 算,美國米諾地爾及非那雄胺的年治療費用分別為 282 美元及 1183 美元。
福瑞他恩治療雄激素脫發研發進展
雄激素性脫發機理:雄激素轉換的二氫睪酮(DHT)與雄激素受體(AR) 的結合過程影響毛囊細胞,造成毛囊阻塞,使其無法吸收營養而萎縮,開 始產生過量掉發。如果沒有及時治療最終導致禿發。福瑞他恩競爭性結合 雄激素受體,減少雄激素與 AR 的結合,從而局部阻止雄激素介導的信號 傳遞。
福瑞他恩治療雄激素性脫發已完成一項中國和美國 Ib 期臨床試驗。試驗顯 示,福瑞他恩安全性良好,無三級以上嚴重不良事件發生。與治療藥物相 關的副作用均為一過性輕度接觸性皮炎,產生原因可能與輔料導致的局部 皮膚過敏有關。此外,福瑞他恩為局部外用制劑,血藥濃度極低,未觀察 到 AR 機制相關的副作用發生。
目前,福瑞他恩治療中國男性雄激素性脫發患者的 II 期臨床已經達到臨床 終點,數據顯示福瑞他恩的有效性及安全性良好,并確定了 III 期臨床的用 藥劑量。2021 年 11 月,福瑞他恩治療男性雄激素脫發的 III 期注冊臨床試 驗獲得 NMPA 批準。這項試驗計劃納入 416 例受試者,試驗時間為 24 周, 主要臨床終點為 24 周結束時目標區域內非毳毛數量(TAHC)對比基線的 變化,目前已經完成首例入組。此外,針對女性雄激素脫發患者的 II 期臨 床試驗也已經完成了首例患者給藥。
福瑞他恩治療痤瘡
尋常痤瘡是一種慢性炎癥性皮膚疾病,以開放或閉合的粉刺及炎性病變 (如丘疹、膿包、或結節)為特點。尋常痤瘡是一種常見疾病,尤其在青 少年人群中更為高發。痤瘡的發病機理涉及多個過程,包括皮脂產生和皮 脂細胞分化、增殖和炎癥。這些過程受循環性激素水平以及局部合成的激 素、神經肽、微生物群、和促炎性細胞因子、脂質介體、抗微生物肽、和 單不飽和脂肪酸(MUFA)的調節。目前的治療方法包括激素藥物(抗雄 激素治療)、局部治療、全身抗生素及異維 A 酸。
中國人群截面統計痤瘡發病率為 8.1%。但研究發現超過 95%的人會有不 同程度痤瘡發生,3%~7%痤瘡患者會遺留瘢痕,給患者身心健康帶來較 大影響。2018 年,中國有超過 1.19 億 10-25 歲的尋常痤瘡患者,預計 2028 年將達到 1.22 億人。美國 2018 年 10-25 歲尋常痤瘡患者約有 3130 萬人,預計 2028 年將達到 3370 萬人。
福瑞他恩治療尋常痤瘡的 I 期臨床試驗已經完成,II 期臨床試驗已經完成首 例入組,公司計劃于 2022 年完成 I/II期臨床并開展 III期臨床試驗。
福瑞他恩銷售預測
福瑞他恩治療雄激素性脫發 II 期臨床已經成功,III 期臨床首例入組,假設 上市成功率為 75%,2023 年上市;雄激素性脫發人群中,使用藥物治療的患者比例逐漸從 2%上升到 5%,福瑞他恩的市場占有率逐漸上升到 20%; 假設福瑞他恩年治療費用為 3000 元,出廠價為終端價的 60%;患者依從 性逐漸從 25%上升到 60%;
福瑞他恩治療痤瘡處于早期臨床試驗中,假設上市成功率為 30%,2024 年上市;痤瘡患者的藥物治療率逐漸從 5%上升到 8%,福瑞他恩的市場占 有率逐漸上升至 5%;年治療費用及出廠價與脫發適應癥一致,患者依從 性從 25%逐漸上升到 50%;
基于以上假設,預計福瑞他恩銷售峰值約為 15 億元,經風險調整后的銷售 峰值為 9.3 億元。
GT20029 治療雄激素性脫發和痤瘡
GT20029 是公司自主研發的 AR-PROTAC 藥物,計劃開發為治療雄激素 性脫發和痤瘡的局部制劑。
蛋白降解靶向嵌合體(PROteolysis TArgeting Chimera,PROTAC)是一 種異型雙功能化合物,其一端是靶向目標靶蛋白的配體,另一端是結合 E3 泛素連接酶的配體,再通過一定長度的 linker 連接。在體內,PROTAC 可 以將目標靶蛋白和 E3 泛素連接酶拉近,從而誘導目標靶蛋白泛素化,然 后降解目標靶蛋白。
GT20029 無法透過皮膚滲透,因此具有不會導致 AR 機制相關的副作用的 優勢。GT20029 還顯示出降解突變 AR 蛋白的潛力,這將有利于治療接受 AR 拮抗劑治療后的患者,同時治療有效的持續時間可能長于 AR 拮抗劑。
2021 年 7 月 28 日,公司宣布 GT20029 中國 I期臨床已完成首批受試者入 組及給藥。2021 年 7 月 13 日,公司宣布 GT20029 已經獲得 FDA 的 IND 批準,將開展治療雄激素性脫發和痤瘡的研究。
ALK-1 抑制劑 GT90001 治療腫瘤
肝癌市場及現有標準療法
中國是肝癌大國,2018 年,中國有 56.17 萬肝癌患者,預計到 2028 年中 國肝癌患者人數將增加至 120 萬人。美國 2018 年肝癌患者約 10.79 萬人, 預計 2028 年將增加至 22.76 萬人。
肝癌藥物市場預計將保持快速增長。2018 年,中國肝癌藥物市場達到 46 億人民幣,預計 2028 年將達到 289 億人民幣。同期美國肝癌藥物市場將 由 9 億美元增長至 46 億美元。
根據 2020 年 CSCO 肝癌指南,晚期肝細胞癌一線的推薦藥物包括索拉非 尼、侖伐替尼、多納非尼、阿替利珠單抗、以及化療等,二線治療的推薦 藥物包括瑞戈非尼、PD-1、和阿帕替尼等。
GT90001 作用機制及研發進展
GT90001 是一種激活素受體樣激酶 1(Activin receptor-like kinase 1, ALK-1)抑制劑。
索拉非尼等抗血管生成抑制劑(VEGF 抑制劑)是目前治療肝癌的關鍵療 法之一。但是部分患者使用 VEGF 抑制劑后會產生耐藥性,從而使 VEGF 抑制劑無法繼續產生療效。激活素受體樣激酶 1(Activin receptor-like kinase 1,ALK-1)在多種腫瘤細胞中選擇性表達。ALK-1 與其配體 BMP9 及 BMP10 結合,調節 SMAD 磷酸化,并促進穩定血管發育。ALK-1 信號 通路的激活可能是造成腫瘤逃避 VEGF 抑制劑的抑制作用的機制之一。 ALK-1 抗體可通過阻止受體結合來抑制 ALK-1 信號通路,從而抑制腫瘤血 管生長及減少血流及血管生成,進而減緩腫瘤生長。
GT90001 是公司于 2018 年從輝瑞引進的一種 ALK-1 人源化單克隆抗體, 是目前國內唯一一個臨床階段的 ALK-1 藥物。公司獲得了 GT90001 在全 球范圍內研發、生產和商業化的獨家權益,授權適應癥涵蓋所有癌種領域。
此前,輝瑞已經在美國及意大利和韓國及日本完成了兩項 ALK-1 單藥治療 晚期實體瘤(包含肝細胞癌)的 I 期臨床試驗。公司正在中國臺灣開展 GT90001 聯合納武利尤單抗二線治療轉移性肝細胞癌的 Ib/II期臨床試驗。
這項臨床試驗為單臂試驗,計劃納入經索拉非尼或侖伐替尼一線治療后疾 病進展或不耐受的晚期肝細胞癌患者。試驗分為劑量遞減的安全性評估階 段和劑量擴展階段。
截至 2020 年 9 月 30 日,在所有 20 例可評估患者中,有 8 例患者達到 PR, 其中 5 例患者達到確認的 PR,確認的 ORR 為 25%(5/20),疾病控制率 為 50%。
安全性方面,沒有觀察到劑量限制性毒性(DLT)。6 例患者報告了與治療 相關的 3-4 級不良反應(30%),其中包括血小板計數降低(n = 3, 15.0%),皮疹(n = 2,10%),天冬氨酸轉氨酶升高(n = 1,5%)。沒有 5 級不良事件的報告。3 例患者(15%)報告了與治療相關的嚴重不良反應 (腎功能不全 G2,肝炎 G2,高淀粉酶血癥 G2)。
與現有晚期肝細胞癌二線推薦療法相比,GT90001 目前的 ORR 數據較好, 期待獲得大規模臨床驗證。
2021 年 2 月,公司宣布 GT90001 單抗聯合納武利尤單抗二線治療晚期肝 細胞癌的全球多中心 II 期臨床的 IND 申請已經獲得 FDA 的批準。2021 年 10 月 9 日,GT90001 聯合納武利尤單抗二線治療晚期肝細胞癌的 IND 申 請獲得 NMPA 批準。
此外,2020 年 7 月,公司與康寧杰瑞達成合作,共同推進 GT90001 聯合 重組人源化 PD-L1/CTLA-4A 雙抗 KN046 治療肝細胞癌等腫瘤的臨床研究。 目前,GT90001 與 KN046 聯合治療晚期或難治性實體腫瘤的 Ib/II 期臨床 試驗已經在中國臺灣啟動,這項試驗計劃納入肝細胞癌、胃癌和胃食管結合處 癌、尿路上皮癌、食管鱗癌等多種晚期或難治性實體瘤患者,2021 年 11 月完成了首例患者給藥。
GT90001 銷售預測
GT90001 治療肝癌目前處于早期研究中,假設上市成功率為 40%,2025 年上市;假設二線肝癌中 ALK-1 藥物滲透率逐漸上升至 6%,GT90001 市 場占有率逐漸從 100%下降到 70%;假設 GT90001 上市時的月治療費用 為 1.6 萬人民幣/月,上市第二年進入醫保后費用為 1 萬人民幣/月(參考二 線肝癌藥物瑞戈約為非尼費用為 1.6 萬/月,阿帕替尼費用約為 1 萬/月), 之后每年下降 2%,出廠價為終端價 70%;患者平均用藥時長參考目前晚 期肝癌二線平均 PFS;
基于以上假設,我們預計 GT90001 在 2030 年的銷售峰值為 4.1 億元,經 風險調整后的銷售峰值為 1.6 億元。
其他研發管線藥物
迪拓賽替:mTORC1/mTORC2 雙抑制劑
迪拓賽替是第二代 mTOR 抑制劑,可同時抑制 mTORC1 和 mTORC2。與 僅抑制 mTORC1 的第一代 mTOR 抑制劑相比,迪拓賽替顯示出更好的治 療效果。
PI3K/AKT/mTOR 的信號途徑有助于調節多種細胞功能,包括細胞增殖、 分化、細胞凋亡及營養。第一代 mTOR 抑制劑對 mTORC2 并無效力,其 可激活 MEK/MAPK 通路及 PI3K/Akt 通路,減輕負回饋抑制,導致降低抗 腫瘤作用。第二代 mTOR 抑制劑迪拓賽替(GT0486)就 ATP 與 mTOR 的催化位點競爭,可以降低 PI3K/mTOR 雙重抑制的毒性,而不影響如 AKT 之類的反饋途徑。
目前,全球還沒有 mTORC1/mTORC2 雙抑制劑獲批上市,在研的 mTORC1/mTORC2 雙 抑 制 劑 中 進 展 較 快 的 是 德 琪 醫 藥 和 新 基 的 Onatasertib(CC-223),目前處于臨床 II 期。公司的迪拓賽替目前處于中 國和美國的 I期臨床試驗中,針對的適應癥為白血病和基底細胞癌。
GT1708F:Hedgehog 信號通路 SMO 抑制劑
髓母細胞瘤和基底細胞癌的發生與 Hedgehog 信號途徑的異常激活有關。 上調 SMO 會激活 Hedgehog 信號途徑。通過抑制 SMO 可以減少小鼠模型 中慢性骨髓性白血病的發生。
目前全球有三款 SMO 抑制劑在美國或歐盟獲批上市,分別是輝瑞的 Glasdegib(急性骨髓性白血病),諾華/Sun 的 Sonidegib(基底細胞癌), 和羅氏的 Vismodegib(基底細胞癌)。在中國,公司的 GT1708F 是第二個 進入臨床階段的 SMO 抑制劑。
GT90008:PD-L1/TGF-β雙抗
公司的 GT90008 具有同時抑制 PD-L1 和 TGF-β的高度活性。通過基因工 程修飾能夠降低其在 CHO 細胞表達蛋白中的降解或片段化,更易于商業 化生產。具有成為同類最佳藥物的潛力。
目前全球尚無 PD-L1/TGF-β雙抗產品獲批上市,目前在研的適應癥包括 非小細胞肺癌、膽管癌、子宮頸癌、以及鼻咽癌。中國在研的 PDL1/TGF-β雙抗中,恒瑞的 SHR-1701 處于較為領先的階段。公司的 GT90008 目前處于臨床 I期研究中。
(本文僅供參考,不代表我們的任何投資建議。如需使用相關信息,請參閱報告原文。)
精選報告來源:【未來智庫】。未來智庫 - 官方網站

劉俊華